



업계에서는 지난해 자문위 내에서도 의견이 갈렸던 ‘렐리브리오’의 승인 사례처럼 치료 옵션이 부족한 루게릭병 환자를 위해 토퍼센 역시 현장 도입에 무게를 두고 있다는 평가가 나온다. 국내 코아스템도 루게릭병 신약 후보 ‘뉴로나타-알’의 미국 내 임상 3상에 박차를 가하면서 기대를 모으고 있다.
|
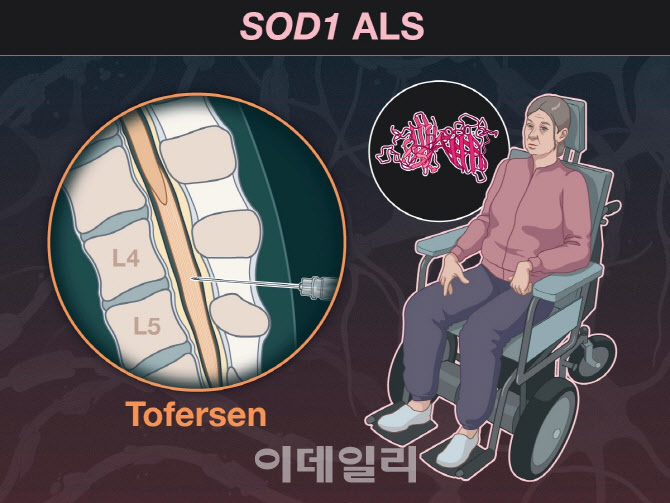
28일 제약바이오업계에 따르면 미국 정부의 퇴행성 신경 또는 뇌질환 신약에 대한 허가 촉진 기조가 이어질 전망이다.
시장조사업체 그랜드뷰리서치에 따르면 대표적인 퇴행성 질환인 루게릭병 환자는 세계적으로 35만 명이다. 이중 약 10%가 미국에 있다. 한국에는 3000명 안팎의 환자가 있는 것으로 확인되고 있다. 이같은 루게릭병 환자를 위한 치료제 시장 규모는 2026년경 8억8000만 달러(한화 약 1조원)에 이를 것으로 전망되고 있다.
지난 22일(현지시간) 바이오젠은 이날 개최된 FDA 자문위 회의에서 ‘토퍼센의 임상적 이점’ 및 ‘토퍼센으로 인한 바이오마커 개선’ 등 두 가지 주제를 논의한 결과, 해당 약물의 가속승인을 지지하는 결론에 도달한 것을 확인했다고 밝혔다.
토퍼센은 ‘슈퍼옥사이드 디스뮤타제(SOD)1’ 유전자 변이로 인한 루게릭병 신약 후보물질이다. SOD1 돌연변이로 인한 루게릭병 환자는 전체(약 35만 명)의 2%인 7500여 명이다. 루게릭병 환자 중에서도 극소수의 유전자 변이 그룹에게 유일한 치료 대안이 될 수 있는 셈이다.
바이오젠에 따르면 토퍼센의 최종 심사 결론 발표를 앞두고 진행된 이날 FDA 회의에서 자문 위원 9명은 토퍼센이 임상적 이점이 있다는데 만장일치로 동의했다. 반면 실질적인 효과에 대한 지표적인 증거 유무에 대한 자문위 투표 결과는 반대가 5표로 많았으며, 찬성과 기권은 각각 3표와 1표를 기록했다.
이런 결과를 종합해 FDA 자문위가 결국 토퍼센의 임상적 이점에 대한 합리적인 가능성이 있다는 데 힘을 실으면서 최종적으로 가속승인을 지지했다는 것이다. 토퍼센은 과거 임상 3상에서 2차 지표인 ‘뇌척수액 내 SOD1 단백질 수치 증가’ 및 ‘미세신경섬유 경쇄’(NfL) 농도 등을 투여용량별로 각각 26~38%, 48~67%씩 감소시키는 효능을 보인 바 있다.
프리야 싱할 바이오젠 연구개발담당 대표는 “신경섬유 감소가 토퍼센의 임상적 이점을 담보할 근거가 된다는 데 의견이 모였다”며 “SOD1 변이 루게릭병 환자 치료에는 가장 핵심적인 기여를 하게 될 것”이라고 말했다.
|

퇴행성 신경질환에 관대한 미국 규제 당국이 렐리브리오의 사례처럼 토퍼센 역시 승인할 것이란 분석이 우세하다. 국내 기업의 미국 내 루게릭병 신약 개발 성공에 대한 기대도 커지고 있다.
코아스템켐온 관계자는 “렐리브리오가 ALS 기능평가 지표를 충족하지 못했음에도 미충족 수요 논리와 데이터 재해석 결과가 맞물려 승인됐고, 토퍼센도 같은 논리다”며 “루게릭병 질환 관련 약물에 대한 FDA의 대응과 그 결론의 방향성을 이런 사례에서 엿볼 수 있다”고 운을 뗐다.
코아스템켐온(166480)은 루게릭병 대상 줄기세포 재생치료제 ‘뉴로나타-알’에 대해 2020년 7월부터 미국 등 글로벌 임상 3상을 진행하고 있다. 지난 2014년 임상 3상을 진행하는 조건으로 국내에서는 뉴로나타 알이 품목허가를 받아 출시된 바 있다. 해당 약물의 국내 시판후 처방 데이터를 분석한 결과 수명연장 효과가 렐리브리오(약 13개월) 대비 4~5배 많은 평균 67개월에 이르는 것으로 분석되고 있다.
앞선 관계자는 “우리 약물의 경우 임상 3상없이 시판됐고, 이제 진행되고 있기때문에 ALS 기능평가 지표에 대한 분석 결과는 아직 나오지 않았다”며 “다만 시판 후 환자에게 실제로 도입된 뒤 나온 결과로 볼 때 뉴로나타-알은 오래전부터 지연제로 쓰이던 경구약 ‘리루졸’, ‘라디컷’을 비롯해 최근에 등장한 렐리브리오나 SOD1 관련 약물 등과 비교해도 수명연장 효과가 월등히 높은 상황이다”고 설명했다. 이어 “미국과 한국 내 임상 3상의 환자 등록이 모두 끝났고, 관련 투여 절차에 박차를 가하고 있다”고 덧붙였다.
코아스템켐온에 따르면 지난 2월 뉴로나타-알의 미국 임상 3상에 최종 목표인원(115명)에 대한 환자 등록도 모두 끝냈다. 회사는 2024년 말 뉴로나타-알의 허가신청서를 FDA에 제출하는 것을 목표로 하고 있다.



![[포토]이가영,부드러운 티샷 공략](https://spnimage.edaily.co.kr/images/Photo/files/NP/S/2024/11/PS24110100330t.jpg)
![[포토] '트릭 오어 트릿' 진행하는 바이든 대통령 부부](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103102211t.jpg)
![[포토] 송민혁 '이글 2개, 버디7개 잡은 날'](https://spnimage.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103100152t.jpg)
![[포토]치솟던 배춧값 대폭 하락…"물량 충분해"](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103101370t.jpg)
![[포토]삼성전자 반도체 영업익 4조](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103101369t.jpg)
![[포토]하모니카 연주가 이윤석의 연주](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103101230t.jpg)
![[포토]민통선 주민들 트랙터 시위](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103101122t.jpg)
![[포토] 서울시예산안 설명하는 오세훈 시장](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103100890t.jpg)
![[포토] 벤틀리모터스코리아, '더 뉴 컨티넨탈 GT 스피드' 공개](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103100418t.jpg)
![[포토] 2024 서울 문화원 엑스포](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/10/PS24103001770t.jpg)
![[포토]이가영,정상을 바라본다](https://spnimage.edaily.co.kr/images/Photo/files/NP/S/2024/11/PS24110100331t.jpg)

![[단독]新폐렴구균 백신 국내 허가…무료접종 판 흔들까](https://image.edaily.co.kr/images/Photo/files/NP/S/2024/11/PS24110100631b.jpg)

